:::
科技新知

由Milasen的開發過程觀察罕見遺傳性疾病個人化寡核苷酸藥物的發展機會與瓶頸
發表日期:2024-05-29
作者:沈哲標(生技中心)
摘要:
本文將介紹針對罕見致命性神經退化性疾病所開發的個人化藥物Milasen的開發經過,它以不到一年的時間內完成疾病分子診斷(突變鑑定)、藥物設計、測試、製造與開始施打於病患,而病患在給藥後一年內有觀察到明顯的症狀療效。
全文:
全球有數千萬兒童患有罕見且可能危及生命的遺傳疾病,這些疾病中有超過7,000種已經對其分子變異有一定的了解。但仍僅有不到10%的罕見疾病具有相對應的治療方法。其原因在於罕病藥物的使用者極少,將造成高額開發成本不易回收,故多數藥廠不會有意願進行相關藥物的開發。而且許多罕病病患具有獨特的基因突變,或發病後的症狀惡化很快,使存活期很短,也無法進行常規的藥物臨床試驗。故這些罕病患者要獲得救治,只能進行量身定製的個人化藥物開發,但多數患者沒有足夠資源來開發客製化藥物,且需要法規配合。本文將介紹針對罕見致命性神經退化性疾病所開發的個人化藥物Milasen的開發經過,它以不到一年的時間內完成疾病分子診斷(突變鑑定)、藥物設計、測試、製造與開始施打於病患,而病患在給藥後一年內有觀察到明顯的症狀療效。本文將概述其開發歷程,及未來若要借鏡這種開發模式進行個人化寡核苷酸藥物時,可能須注意的機會與瓶頸,期能對未來有志於開發個人化療法者產生拋磚引玉的效果。
一、反義寡核苷酸藥物可針對特定標靶進行治療,現有多項產品上市
反義寡核苷酸(Antisense oligonucleotide, ASO)為短鏈單股合成的核酸(由DNA、RNA或多種人為合成核苷酸組成)序列,長度大約介於15~25個核苷酸之間,序列通常有經過化學修飾。ASO可與特定的標靶RNA序列(如mRNA等)利用鹼基配對完全互補形成雙股結構,進而影響基因的蛋白質表現(如產生基因沉默)、促使目標RNA被抑制轉錄等作用。統計至2024年Q1為止,目前已有12個ASO藥物上市(表1),所治療的主要適應症為罕見疾病如:裘馨氏肌肉萎縮症(Duchenne muscular)、遺傳性澱粉樣蛋白疾病(hereditary transthyretin-mediated amyloidosis, hATTR)、與神經元蠟樣脂褐質儲積症(Neuronal Ceroid Lipofuscinosis 7, NCL)、脊髓性肌肉萎縮(Muscular dystrophy spinal, SMA)與同型合子家族性高膽固醇血症(homozygous familial hypercholesterolemia, HoFH)等。
表1 已上市反義寡核苷酸藥物(ASO)
| 產品名稱 |
藥物名稱 |
公司名稱 |
首次核准 |
適應症 |
| Vitravene |
fomivirsen* |
Ionis/ Novartis |
1998 |
巨細胞病毒性視網膜炎(cytomegalovirus retinitis, CMV retinitis) |
| Kynamro |
mipomersen |
Sanofi |
2011 |
同合子家族性高膽固醇血症 |
| Defitelio |
defibrotide |
Gentium |
2013 |
肝靜脈阻塞性疾病(hepatic veno-occlusive disease, VOD) |
| Exondys 51 |
eteplirsen |
Sarepta |
2016 |
裘馨氏肌肉萎縮症 |
| Spinraza |
nusinersen |
Biogen/ Ionis |
2016 |
脊髓性肌肉萎縮 |
| Tegsedi |
inotersen |
Ionis |
2018 |
遺傳性澱粉樣蛋白疾病 |
| Milasen |
milasen |
Boston Children’s Hospital |
2018 |
神經元蠟樣脂褐質儲積症 |
| Waylivra |
volanesorsen |
Ionis/ Akcea |
2019 |
高三酸甘油脂血症、脂蛋白脂肪酶缺乏(Hypertriglyceridemia, lipoprotein lipase deficiency) |
| Vyondys 53 |
golodirsen |
Sarepta |
2019 |
裘馨氏肌肉萎縮症 |
| Viltepso |
viltolarsen |
NS Pharma |
2020 |
裘馨氏肌肉萎縮症 |
| Amondys 45 |
casimersen |
Sarepta |
2021 |
裘馨氏肌肉萎縮症 |
| Wainua |
eplontersen |
Ionis |
2023 |
遺傳性澱粉樣蛋白疾病 |
註*:全球第一個ASO藥物,後因病患人數少、用量少、於2003年下市
資料來源:Citeline;DCB產資組ITIS研究團隊(2024.04)
二、Milasen的簡要開發歷程
米拉(Mila makovec)出生於2010年11月,在她3歲時,父母發現她的右腳開始有內翻的現象,5歲時因語言能力不佳、社交退化及活動靈敏度不良的情況日益嚴重而前往就醫,滿6歲前的幾個月中,其症狀已迅速發展到如:視力喪失、頻繁的跌倒、發音障礙與吞嚥困難等症狀,因而開始住院。磁振造影顯示出米拉有輕度的小腦萎縮,24小時腦電圖也顯示一天內會發生數次的亞臨床癲癇(subclinical seizures)發作。最終在皮膚切片中發現異常、高電子密度的溶小體包涵體(lysosomal inclusions)在螺旋指紋狀(swirling fingerprint pattern)特徵中出現,因而病患被診斷為巴頓氏症(Batten’s disease)。
巴頓氏症是一種致命的神經系統代謝疾病,通常病發於幼兒期。巴頓氏症是已知有13種類型的神經元蠟樣脂褐質儲積症(Neuronal ceroid lipofuscinoses, NCLs)的通稱。它是由十多個各式CLN基因(如CLN1、CLN2等)中任一個基因的突變所導致。此外,由於巴頓氏症屬隱性遺傳疾病,故幼童染色體中必須有兩套CLN基因突變拷貝(copy)(分別來自父母)才會發病。這些突變影響到細胞分解與清除胞內廢物的能力,故造成異常蛋白質、糖類和脂質的堆積,進而使視網膜與中樞神經系統中的神經細胞死亡產生病變,病患會有失明、癲癇、認知(思考和推理)、無法行走或與人互動等症狀,最終導致病患的死亡。且通常患者的症狀出現得越早,壽命就越短。
米拉在最初已在MFSD8(Major facilitator superfamily domain containing 8)基因(亦稱為CLN7)上發現一個突變,但尚未發現第二個。由於波士頓兒童醫院的臺裔醫師游維文(Dr. Timothy Yu)的團隊專精於全基因體定序(Whole genome sequencing, WGS),主動向家屬表示願意協助解答迷團:找到第二個突變位點。團隊在取得患者父母的書面知情同意書,與波士頓兒童醫院的機構審查委員會核准的人類受試者研究方案(human subjects research protocol)後,取得血液和皮膚的檢體用於基因定序與分析。團隊最終發現在MFSD8基因上另有一個SVA(SINE-VNTR- Alu)逆轉錄轉座子(retrotransposon)的插入,造成基因的不正常剪接(Splicing)與序列錯誤連接,將導致蛋白質轉譯作用過早中止。
在發現第二個基因突變位點及影響後,團隊聯想到在2016年獲FDA核准應用於治療脊髓性肌肉萎縮症(Spinal muscular atrophy, SMA)的ASO藥物Spinraza(nusinersen),該藥物之作用機轉為改變SMN2 RNA的錯誤剪接模式,而SMA與米拉所罹患的巴頓氏症的疾病類型與致病機轉相彷,故推測運用ASO作為藥物亦可能有助於修正錯誤的RNA剪接機致,並恢復患者體內正常的MFSD8基因表現。團隊著手設計可用的ASO序列與進行合成,並使用患者的纖維母細胞(fibroblasts)進行測試。其中發現了三種可將「正常:突變體剪接比(normal:mutant splicing ratios)」提高2.5至3倍的ASO藥物,其中編號TY777的ASO在試驗中最具療效,故被選為先導候選藥物,並依病患米拉的名字命名為「Milasen」。Milasen是由22個核苷酸所組成的ASO藥物,其設計上延用與Spinraza相同的主鏈和糖化學修飾。
在Milasen藥物的體外試驗部分,根據細胞RNA定序結果,來自患者的纖維母細胞中加入Milasen後,其正常剪接量相較未給藥的組別增加了三倍之多。且細胞中與溶酶體功能障礙(lysosomal dysfunction)相關的細胞表型特徵在給藥後獲得減少,顯示藥物具恢復細胞正常功能的效果。此外,Milasen的序列經分析後顯示,在人類基因組中產生脫靶結合的可能性不高。為了評估Milasen在動物體內的安全性,大鼠以髓鞘內注射藥物的方式進行毒理試驗。數據顯示在特定劑量下,並未在動物上觀察到不良反應。同時,團隊亦委託相關公司進行臨床試驗所用Milasen原料藥的製造與配製。
由於米拉的病情持續惡化,與考慮到患者越晚進行治療則臨床預後可能越差。相關細胞與動物數據及研究計畫在經過科學與倫理審查後,根據「擴大近用(Expanded Access)」機制,團隊向FDA申請了研究新藥申請(Investigational New Drug application, IND)的許可以啟動臨床研究性治療計畫,在動物毒理學試驗開始1個月後,團隊獲准在病患上啟動Milasen的臨床研究(圖1)。由於Milasen與已上市的Spinraza的長度相彷、具相同化學修飾、應用於相同的中樞神經組織中,故本次Milasen的臨床治療方案以nusinersen的給藥方式為範本,並以脊髓鞘內注射給藥。給予病患的起始劑量為3.5 mg,約每2週增加7mg,最終達到42 mg(約開始給藥的第82天)。劑量遞增至42mg後,再額外給予兩次給藥後,約每3個月進行一次維持劑量( maintenance dosing)的施打,直到給藥後第301天進行最後一次藥物施打。
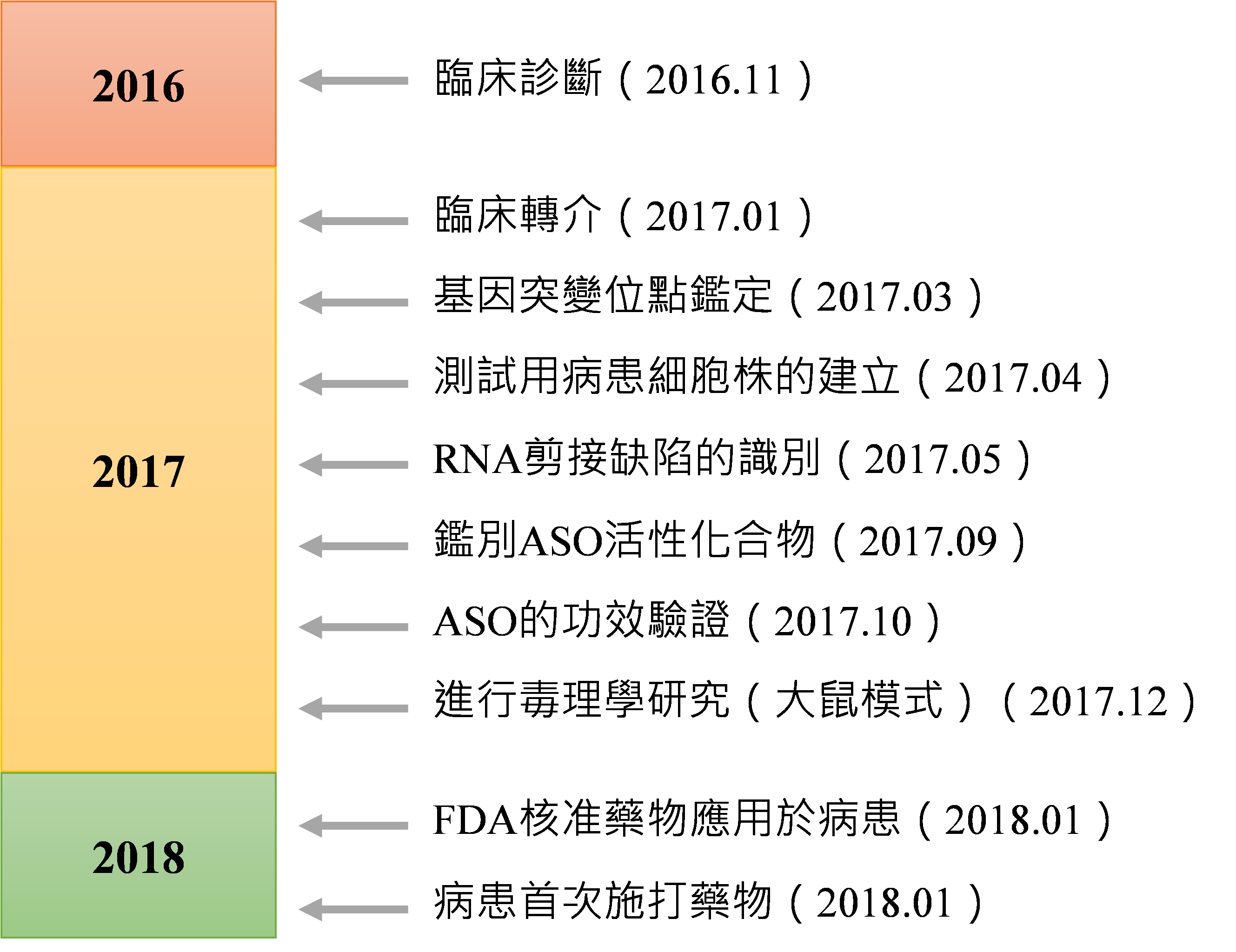
資料來源:N Engl J Med. 2019;DCB產資組ITIS研究團隊(2024.04)
圖1 Milasen寡核苷酸藥物從完成診斷到首次人體給藥的簡要歷程
三、Milasen個人化療法臨床試驗的早期可看到症狀惡化減緩,但無法逆轉病程的發生
在安全性方面,治療的第一年中,並未觀察到嚴重不良事件或生命徵象的不良反應。在癲癇發作方面,在原有給予的抗癲癇藥物治療方案不變之下,臨床試驗期間給予病患Milasen藥物後,患者每天的癲癇發作頻率由15至30次減少至每天0至20次。每次持續時間由1至2分鐘減少至不到1分鐘。顯示癲癇發作的頻率和持續時間均減少了50%以上。在頭部磁振造影監測部分,影像顯示病患腦容量在Milasen治療開始後的7個月仍持續減少,與過去3年在病患身上所觀察到的趨勢相同。在其它進行巴頓氏症(如CLN5型)基因治療的動物試驗中也發現,在症狀出現後才進行治療則僅能減緩症狀惡化,但無法抑制腦容量的持續減少。在運用Vineland 適應性行為量表(Vineland-II)進行神經疾病學與神經心理學的評估方面,在試驗第6天和第100天之間,11個神經學和神經心理學的子評分指標(subscores)中有7個下降,顯示患者在日常生活技能與溝通社交上殘存的適應性能力仍在持續喪失。然而Milasen仍屬於研究用藥物,且不適合用於治療其他巴頓氏症的患者,因為它的藥物設計是根據患者的特定突變序列來量身定製的,不過其開發過程、協助資源、援引的法規與待改善的瓶頸的資訊對個人化ASO藥物的開發仍具有極高的參考價值。
四、病患使用Milasen或類似模式開發藥物可能產生未如預期的結果
病患米拉在2018年1月底(年齡約7歲2個月)開始接受個人化藥物的試驗性治療,在接受試驗前已幾乎無法行走、語言能力減退、癲癇頻繁發作,並需要使用鼻胃管來進食與喝水。給予藥物後雖然癲癇症狀在初期有獲得改善,且能再次使用嘴巴進食,但事實上,給藥並沒有使米拉痊癒,其反應仍然遲緩。在給藥數年後,病情又開始緩慢的惡化。米拉最終仍於2021年2月11日去世(享年10歲3個月),自接受Milasen藥物治療後又存活了3年。一般認為,米拉在病程的晚期(文獻中CLN7型巴頓氏症患者的平均發病年齡為3.3歲)才開始治療可能是造成療效有限的因素之一。
然而個人化療法或ASO藥物治療本身仍存有許多未知的變因。波士頓兒童醫院的同一臨床研發團,在後續以相近模式針對2位具有相同的罕見KCNT1基因突變的患者開發了個人化ASO藥物Valeriasen。第一位患者在2020年9月接受藥物脊髓注射後,症狀在治療的初期有獲得改善。但在施打後11個月發現腦積水(hydrocephalus),1個月後即去世。第二位患者在2021年6月接受給藥,其癲癇發作頻率在給藥後初期有明顯下降,惟同年8月亦診斷出有腦積水,但停藥後癲癇發作率即恢復上升。雖然過去在施打實驗性或已上市ASO藥物的患者身上均曾有發生腦積水的案例,但目前仍無法確定此一症狀是由藥物所引起,或是病患原有的神經系統疾病所導致,而這也是目前非商業化開發個人化藥物的風險所在:受試者極少且其臨床試驗數據不需於研究社群中公開,使得同一類試驗的安全性風險無法事先被預估。故建立起客製化藥物的臨床試驗資訊的分享機制,亦是使此一新興領域朝正向發展的重要基礎。
不過,綜合而言,面對異常嚴重或危及生命的罕病患者在臨床時程的緊迫性,臨床團隊與病患與家屬皆需認知此類藥物在開發與使用上仍具有極大風險。但ASO藥物具有可根據病患突變序列特異性作量身定製的特性、藥物在中樞神經系統中具有良好吸收與長半衰期、ASO藥物製造的相對簡單性(與其他大小分子藥物相比)使其能在短期內完成小批量生產等優勢特性,ASO藥物在罕見疾病的個人化藥物領域仍屬極具潛力的開發平台。
五、本案後續效應與影響力
為了規範未來此類特殊個人化ASO藥物的臨床試驗需求,美國食品暨藥物管理局(Food and Drug Administration, FDA)於2021年1月公布給研究申請者的「個人化反義寡核苷酸藥品的IND提交:行政和程序建議(Investigational New Drug Submissions for Individualized Antisense Oligonucleotide Drug Products : Administrative and Procedural Recommendations)」指引草案,並於4月接續發布了「用於嚴重衰弱或危及生命的疾病之個人化反義寡核苷酸藥品的非臨床試驗(Nonclinical Testing of Individualized Antisense Oligonucleotide Drug Products for Severely Debilitating or Life-Threatening Diseases Guidance for Sponsor-Investigators)」指引草案。在試驗藥品的製造管制部分,2021年12月也進一步公布「用於治療嚴重衰弱或危及生命疾病之個人化反義寡核苷酸藥品於臨床試驗申請案的化學製造與管制建議(Investigational New Drug Application Submissions for Individualized Antisense Oligonucleotide Drug Products for Severely Debilitating or Life threatening Diseases: Chemistry, Manufacturing, and Controls Recommendations)」指引草案以完備相關的申請規範。
觀察非臨床試驗指引的內容,可初步了解法規單位對此類與時間賽跑的案件的申請要求與和常規案件的差異。其中如:(1)相較於非罕病治療藥物,由於適用指引的病患較少,故所要求用於支持首次人體(First-in Human, FIH)臨床試驗申請的臨床前安全性資料將會較少。但必須出具非臨床的體外和/或體內概念驗證(proof of concept, POC)數據,以支持此一個人化ASO試驗藥品對受試者之可能效益;(2)臨床試驗申請案應包含非臨床安全性資料如:脫靶評估(hybridization-dependent off-target assessment)、一般毒性試驗、安全性藥理學等,基因毒性試驗報告則為非必需項目;(3)申請案應藉由3個月的毒性試驗完成對合適人體起始劑量、劑量遞增模式的長期安全性的評估。若適應症為臨床表型進程緩慢的案件,須提供3個月期毒性試驗的完整報告。若案件疾病的臨床表型屬嚴重不可逆或將快速進展至死亡,則可先提供正在執行的動物毒性試驗(3個月)內至少2週的存活期間數據(in-life data),且應儘快提出該試驗完整的報告初稿;(4)計畫中的FIH臨床試驗起始劑量,應根據現有數據確認具有預期的藥理作用且具安全性。而人體使用的最高劑量,應由受試者的臨床情況、臨床與非臨床資訊作綜合判斷。
此外,本案例亦在英國產生對個人化罕病藥物趨勢與可行性的推動正面的效應。由於英國擁有全世界規模最大的基因體數據庫(UK Biobank),且持續的投資使英國已位於全球基因體學的最前端。早在2020年即規劃要以10年的時間,建立起世界上最先進的基因體醫療保健系統,且著重於三個領域:(1)運用基因體學進行早期發現與預防;(2)基因體診斷技術與個人化醫療;(3)基因體學的應用與突破性研究。由於推行數十年針對新生兒的傳統足根採血(heel prick test),不但耗時且可能無法篩檢出許多先天性疾病,進而對新生兒進行早期診斷與治療,以改變孩童的命運。2023年下半年已啟動一項名為Genomics England的計畫,針對10萬名新生兒的全球首例國家級WGS計畫,預計將篩檢200種罕見但可因早期治療而獲益的遺傳疾病,且能在檢測後2週內即可得知結果,並藉此研究開發罕病的檢測與治療方法。當前進行WGS計畫所需的成本與時間已顯著減少,且已知分子機轉的疾病數量持續增加,故基於新的基因資訊而開發的新藥與療法研究數量也在穩定增加。雖然相關案例如Milasen顯示建立專供單一罕病患者使用的ASO療法具可執行性,但因為大多數患者並沒有資源與途徑開發或取得這類藥物,故藉由政府或私人的資源建立起個人化藥物的可運作模式與平台,將是一個重要且能對全球罕病患者產生重大影響的起點。
此外,英國因觀察到Milasen等個人化藥物的開發案例可為患有罕見遺傳疾病的兒童及其家人帶來巨大的改變。英國政府在2023年11月宣布啟動一項為罹患罕見疾病的兒童提供個人化療法的計畫:Rare Therapies Launch Pad。由此一前瞻計畫的參與單位即可了解其能量與願景,成員包括:Genomics England計畫、英國藥物及保健產品管理局(Medicines and Healthcare products Regulatory Agency, MHRA)、開發罕見疾病藥物的牛津-哈靈頓罕見疾病中心(Oxford-Harrington Rare Disease Centre)、推動Milasen藥物開發的米拉奇蹟基金會(Mila's Miracle Foundation)與英國製藥產業協會(Association of the British Pharmaceutical Industry, ABPI)。這個計畫的執行面向將涵蓋:疾病診斷、療法設計、藥物快速製造、治療方案,也將同步收集有助於確認個人化療法效益的各種數據。其最終目標將致力於為英國數十萬患有罕見且可能危及生命的遺傳性疾病的兒童,取得快速、安全的個人化療法與適用的法規監管路徑,同時將相關經驗在英國建立起具延續性與拓展性的模式。這是一項世界領先的嘗試,旨在建立一條「新的簡化監管途徑(new streamlined regulatory pathway)」,以允許設計和使用「一次性藥物(one-off drugs)」。這項計畫推動後的首個項目即為:研究運用個人化ASO藥物來治療罹患具致命性罕見腦部疾病的兒童。當然Rare Therapies Launch Pad也有助於確保英國位於罕病治療領域與創新法規監管上的前端,並鞏固其在基因體學應用領域的全球領導地位。
六、結論
綜合來說,一項個人化藥品要能成功完成開發且具有長期療效與安全性,先決條件是具備充足的經費與資源,及紮實的科學驗證數據等重要條件。在驗證數據方面,由於個人化療法的開發過程中受試驗的人數極少,故能獲得關於藥物在人體中的反應數據也有限。同一開發模式未來要能廣泛的應用於同病症其他病患的個人化藥物開發,還需收納更多個案的數據來調校開發方法與設定,以減少未知副作用與增加療效。然而,增加更多臨床案例數據之外,速度對於致命罕見疾病療法的開發也極為重要,開發者與法規單位須在藥物安全性的要求與開發速度間取得平衡。在經費方面,能應用至人體的個人化藥物的開發與生產所需經費必十分龐大且藥價高昂,因罕病個人化藥物非屬大量商業生產的產品,無法將成本分攤。以本案為例,米拉的母親在女兒6歲被診斷出患有無法治癒的致命巴頓氏症後,她成立了米拉奇蹟基金會(Mila's Miracle Foundation, MMF),基金會從6,000多名善心支持者籌集了近500萬美元的善款,以支持新的疾病療法的開發與協助病友(基金會在米拉過世後仍持續運作)。顯見未來若要啟動個人化藥物開發計畫,須找到對經費支付端(政府資源、基金會、保險公司、私人捐贈等)與病患端皆能獲益的模式,才有機會將此開發模式作長遠的推廣。
除了藥物開發之外,生產端也需要配合,個人化藥物的生產特性包括:(1)每批次生產量極小;(2)每批次須快速生產;(3)總生產批次須提高。因此,具備少量多樣的彈性客製化藥品(如核酸藥物)生產能量的CDMO公司是必需的,這類公司能商業獲利運作的先決條件擁有藥物自動化生產裝置平台,以簡單、低成本、高速與標準化的模式增加個人化藥品的可靠度。目前國內已有廠商布局ASO的生產,如臺灣生物醫藥製造、松瑞製藥等公司,但尚未擴及個人化藥物生產,未來若隨相關產業發展越來越成熟,市場機制漸漸明朗,相信將會有廠商看到此處的發展機會。
至於要能快速的將實驗用藥物於個人上進行人體治療試驗,找到相對應的適用法源依據更是核心關鍵。雖然我國目前尚未有個人化罕見疾病ASO藥物的開發案例,亦無相關的藥物開發指引。但為因應國際趨勢,我國的醫藥品查驗中心(Center for Drug Evaluation, CDE)參酌先進國家作法,亦於112年7月公布「用於治療嚴重衰弱或危及生命疾病之個人化反義寡核苷酸藥品於臨床試驗階段之化學製造管制指導原則」,且CDE已預估在今年內會公布此類藥品的非臨床藥毒理研發策略指導原則。故法規單位已逐步建立起可供製藥業者遵循的方向,為我國未來發展造福罕病患者的個人化ASO藥物奠定重要碁石。而在醫療緊急使用的法規方面,各國政府對於醫藥品的臨床使用和上市都會進行嚴格監管措施,以避免國民健康受到未知風險的傷害。以Milasen開發案的美國為例,法規中適用於管理醫療緊急使用需求的機制主要有兩類,第一類為針對個案性或小規模的臨床緊急需求的「擴大近用」機制,第二類為因應公共衛生緊急事件,欲發展醫療反制措施所訂立之「緊急使用授權(Emergency Use Authorization, EUA)」機制。其中本文所述Milasen開發案例所援引使用的「擴大近用」機制指主管機關可在特定情形下同意,將尚未經過查驗登記許可的藥物、生物製劑或醫療器材於臨床上使用,其目的為因應「臨床需求」與「維護國民健康」這兩個目的產生矛盾的時機,而「擴大近用」機制通常也被稱為「恩慈使用(compassionate use)」。
我國對於醫療緊急使用授權的機制,則分別在《藥事法》第48條之2、《醫療器材管理法》第35條中規定,並在相關子法:《特定藥物專案核准製造及輸入辦法》或《特定醫療器材專案核准製造及輸入辦法》加以補充。目前我國相似於美國的「擴大近用」的可行機制,即所謂的恩慈條款,可見於前述「特定藥物專案核准製造及輸入辦法」所闡明的:為預防、診治目前國內尚無適當藥物或替代療法之危及生命或嚴重失能之疾病,或為因應公共衛生需求之緊急情事,而有必要使用尚未取得許可證之藥物,藥事法中已明定未取得許可證之藥物,得由特定醫院為特定病患向中央衛生主管機關專案申請製造或輸入。
此外,客製化藥品的臨床試驗委託者的界定,與投保責任保險的特殊機制也需要釐清。在藥品優良臨床試驗準則第47條中規定:「試驗委託者應負責試驗主持人或試驗機構因試驗所生之賠償責任或投保責任保險。但因試驗主持人或試驗機構之醫療疏失者,不在此限」。由於國內尚無類似客製化藥品開發與試驗案例,關於藥物臨床試驗責任保險,須預先探討相關可行性,也將有助於我國未來精準醫療上更具完整性的發展。由於客製化罕病藥物的臨床試驗人數與相關資訊極少,根據國外的經驗,及早建立起兼顧個資法規的藥物臨床開發數據的研究社群分享機制,亦是使此一新興領域朝正向發展的重要基礎。
綜合上述,Milasen的開發案例在結合:美國的「擴大近用」機制、成熟的WGS技術以快速找出突變位置、參考先前類似疾病領域的ASO藥物的開發與治療模式、ASO可快速設計與生產的優勢、找到符合臨床試驗用藥規範的ASO生產公司、與具醫療與臨床試驗能量的團隊等綜合因素下,才能促成本案在極短時間內就完成診斷與個人化罕病藥物的設計、生產與治療流程。
雖然最後唯一的用藥病患仍因為不可逆轉的疾病退化病程,或太晚開始進行治療等因素下,仍然不敵病魔而過世,但我們從Milasen的案例中可觀察到各種有助於加速療程開發的條件、學習到首次個人化的罕病ASO藥物開發與臨床應用經驗、結合早期診斷罕病與早期治療可能產生的效益、結合基金會/臨床研究者/試驗用藥生產商/法規/政府資源的罕病個人化藥物開發模式,相信能提供給國內產學醫界未來推動個人化罕病療法一些啟示。Milasen的開發案是一則感人但留有缺憾的故事,若能借鏡與修正本案例的經驗、整合各種資源以推動罕病個人化療法的發展,相信將能更大的彰顯米拉的生命價值與意義。。
(本文作者為生技中心執行產業技術基磐研究與知識服務計畫產業分析師)
 點閱數
點閱數:
632
